1月7日,金沙9001w以诚为本入口基础医学院孙金鹏教授研究团队与浙江大学张岩教授团队、中国科学院上海药物研究所徐华强研究员团队精诚合作,全力攻关,在金沙9001w以诚为本入口易凡教授团队和于晓教授团队的协助下,在Nature在线发表了题为“Structures of glucocorticoid-bound adhesion receptor GPR97-Go complex”最新的研究成果。金沙9001w以诚为本入口基础医学院博士生平玉奇、肖鹏副教授、硕士研究生赵儒嘉、浙江大学基础医学院博士研究生毛春友和上海药物所蒋轶研究员为本文共同第一作者;孙金鹏教授、张岩教授、徐华强研究员为共同通讯作者,金沙9001w以诚为本入口易凡教授、于晓教授为论文共同作者。

该研究首次鉴定并解析了糖皮质激素与其膜受体GPR97结合的复合物电镜结构,这也是世界上首次解析的黏附类GPCR在配体激活情况下与G蛋白形成复合物的电镜结构。
黏附类G蛋白偶联受体(Adhesion G protein-coupled receptors, aGPCRs)在GPCR超家族中是一类进化上比较古老的亚群,它又可以分为8个小的亚群,总共33种GPCR蛋白,其中大部分成员都是孤儿受体。aGPCRs在生物体许多重要的生理过程中起到关键分子开关的作用,比如脑的发育、水盐调节、炎症以及细胞命运决定等。很多黏附类受体的突变与多种人类疾病相关,比如VLGR1的突变和耳聋密切相关,而GPR64的突变和男性不育相关,但这些突变如何导致人类疾病,是否可以针对这些黏附类受体开发出相应的药物尚不清楚。与GPCR超家族各成员相比,aGPCR除了具有7次跨膜核心(7TM)之外,在拓扑结构上具有明显的特征。aGPCR胞外区域特别长,并且存在具有不同功能的结构域,目前领域里普遍认为aGPCR通过结合胞外的基质蛋白或者可溶性小分子被激活,然而是否有小分子配体直接可以直接激活7次跨膜核心尚不清楚。
从2010年回国建立实验室开始,孙金鹏教授课题组很早便对aGPCRs这样一类重要的细胞膜受体展开了研究。早期的团队研究发现,听力相关的黏附类受体VLGR1 (GPR98) 6236位移码突变使受体丧失结合具有Gi通路活性的PDZ7的能力,进而导致VLGR1下游Gi通路持续激活,是导致听觉失常的重要原因[1]。在雄性生殖系统中,GPR64能够通过Gq和Arrestin在非纤毛细胞的顶膜与离子通道CFTR的偶联,调控附睾组织中输出小管的水盐代谢和重吸收,进而对雄性生殖起到了重要作用[2]。除了以上两个受体之外,孙金鹏教授课题组还与易凡教授课题组合作发现Gpr97缺失抑制HuR蛋白表达,导致Sema3A mRNA稳定性降低以及Sema3A蛋白表达降低,从而缓解IRI引起的肾功能损伤[3]。
为进一步寻找孤儿受体GPR97的配体,研究团队通过定向筛选的方法,发现糖皮质激素及糖皮质激素类药物可以高亲和力的结合GPR97,并激活GPR97. 糖皮质激素(GC)是机体内极为重要的一类调节分子,它对机体的发育、生长、代谢以及免疫功能等起着重要的调节作用,是机体应激反应最重要的调节激素,也是临床上使用最为广泛而有效的抗炎和免疫抑制剂。早在1950年,美国的菲利普·肖瓦特·亨奇、爱德华·卡尔文·肯德尔和瑞士的塔德乌什·赖希施泰因便因为发现和应用糖皮质激素治疗风湿性疾病而获得了诺贝尔奖。但前人大量的研究显示,糖皮质激素的作用模式,主要是通过与其核受体(GR)结合,并穿过核孔,在细胞核内调控相关基因的表达而发挥作用,这种作用方式一般需要较长的时间才能起作用,被称为基因组机制。但其实,科研人员很早便发现,糖皮质激素也能够通过快速的方式引起细胞和机体的变化。比如,中科院院士、神经生理学家陈宜张先生,早在20世纪80年代便发现,糖皮质激素能够在2分钟内使豚鼠神经节神经元(ganglion neurons)发生膜电位超极化,也能在PC12细胞中快速的抑制nicotine引起的钙离子流[4-7]。这提示我们,生物体内可能存在着糖皮质激素的膜受体,它们能够介导糖皮质激素的快速反应。另外,有研究发现糖皮质激素的快速反应与G蛋白有密切关系,Gi的抑制剂PTX能够抑制糖皮质激素的快速作用,因此,研究人员推测,糖皮质激素的膜受体很可能是GPCR。然而,这四十年以来,糖皮质激素的膜受体一直未能真正确证,糖皮质激素的快速作用的机制依然成谜。
前期已有研究报道,治疗哮喘药物丙酸倍氯米松(BDP)能够在体外激活GPR97。通过结构观察,研究团队们发现BDP是糖皮质激素类药物倍氯米松(BCM)的衍生物,在17和21位有两个丙酸的修饰。这两个丙酸的修饰在激活GPR97过程中是否必须,前期的文章中并没有给予说明。团队于是怀疑两个丙酸的修饰可能不是必须的,而BCM以及其他的糖皮质激素类化合物都可能是GPR97的配体。研究团队于是对内源性激素进行了系统的筛选,最终发现糖皮质激素类的氢化可的松(cortisol)、可的松(cortisone)以及11-脱氧皮质醇(11-deoxycortisol)都能够激活GPR97。此外,地塞米松(Dexamethasone)具有更强的GPR97激活能力。进一步的实验显示,氢化可的松和可的松都能够通过GPR97明显抑制forskolin诱导的cAMP上升。而通过Gqo解离实验,研究人员确认,配体作用于GPR97后最终激活了Go信号通路。
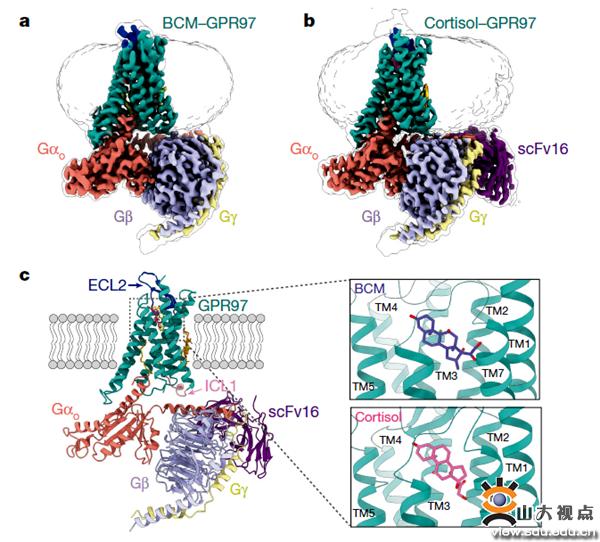
图1. GPR97结构
接下来,研究人员采用单颗粒冷冻电镜分别对外源配体倍氯米松(BCM)以及内源性配体氢化可的松(cortisol)激活GPR97后形成的复合物进行了结构重塑。我们应用单颗粒冷冻电镜技术解析了GPR97在配体激活情况下与G蛋白的复合物结构。其中倍氯米松与GPR97-Go复合物结构的分辨率为3.1Å,氢化可的松与GPR97-Go复合物结构的分辨率达到了2.9Å,如图1所示。
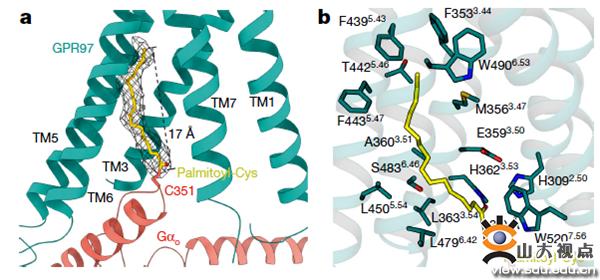
图2. Go棕榈酰化修饰
从电镜结构中我们可以发现,与其他已经解析的GPCR结构相比,GPR97的七次跨膜螺旋呈现独特的空间分布,其螺旋的长度与其他受体也有很大的不同;以前的研究表明,GAIN结构域和七次跨膜结构域在激活GPCR的过程中是作为一个一体的核心来发挥作用的,但出乎意料的是,研究人员在结构中发现糖皮质激素结合到了GPR97七次跨膜正构结合位点口袋(orthosteric binding pocket)中, 配体结合口袋狭长,呈椭球形,主要通过疏水作用力和氢键与受体结合;GPR97激活机制比较特殊,和classA家族受体相比,没有保守的PIF motif、DRY motif和NPxxY motif,GPR97首先通过toggle switch W6.53识别配体,激活的受体借助新发现的upper Quaternary core(UQC)将受体TM3-TM5-TM6捆绑在一起,然后通过HLY motif介导与G蛋白结合。从胞内侧看,受体七次跨膜螺旋组成较大的张开口袋,3个胞内环都参与了受体与G蛋白的相互作用,胞内环与受体的组成性激活非常相关,某些重要氨基酸突变,能显著改变受体的自激活能力。
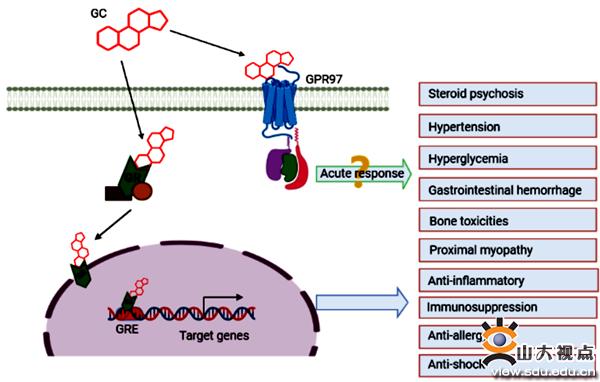
图3. GPR97功能
不仅如此,电镜结构还发现在Gαo的α5 helix C351位点处存在棕榈酰化修饰,这是GPR97-Go复合物中特有的,在其他已有结构的GPCR-Gi/Go复合物中未发现此修饰,以与Gi结合的D2R(多巴胺受体)为对照,将Gα C351位氨基酸突变,结果发现突变只影响GPR97与G蛋白偶联,不影响多巴胺受体与G蛋白偶联。
综上所述,研究人员首次发现了糖皮质激素的高亲和力膜受体,并通过单颗粒冷冻电镜技术解析了黏附类GPCR家族中GPR97在糖皮质激素的激活作用下与G蛋白的复合物结构,从而在原子分辨率上详细阐释了糖皮质激素如何和膜受体作用,激活该受体,并与G蛋白偶联的清晰过程。该成果无论是对于糖皮质激素膜受体功能研究还是黏附类GPCR的激活机制理解都起到了很重要的推动和示范作用。
金沙9001w以诚为本入口基础医学院孙金鹏教授研究团队在GPCR跨膜信号转导领域连续取得进展。2015年提出磷酸化编码的笛子模型理论(Nat Commun. 2015Sep8;6:8202.),进一步揭示了GPCR磷酸化编码别构调控SH3 domain蛋白的多聚脯氨酸码头分选机制(Nat Chem Biol. 2018Sep;14(9):876.);2017年发现多种GPCR与离子通道偶联的新机制以及介导心血管事件(Nat Commun.2017Feb9;8:14335.;Nature Commun.2018Jan2;9:11.;Elife. 2018Feb2;7:e33432.);2020年利用冷冻电镜技术揭示胆汁酸受体配体识别及激活的独特机制(Nature.2020Nov;587(7834):499-504.),并发展新的一维氢谱探针,证明配体对Arrestin的调控可以不依赖于对GRK的选择(Nat Commun. 2020Sep25;11(1):4857.)。
以上研究得到了国家杰出青年科学基金和国家自然科学基金的资助与支持。
文章链接:https://www.nature.com/articles/s41586-020-03083-w








