8月5日,基础医学院孙金鹏教授研究团队和上海交通大学医学院、宁夏医科大学张健教授和陆绍永教授研究团队通力合作,在Nature Communications杂志(2021年IF=14.919)在线发表了题为“Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design”的研究论文。中科院上海药物研究所博士生何欣恒、金沙9001w以诚为本入口基础医学院助理研究员杨照和硕士研究生周淑华为本文共同第一作者,孙金鹏教授、张健教授和陆绍永教授为共同通讯作者。
G蛋白偶联受体(GPCR)是人类基因组中最大的膜蛋白家族,参与调控人体中大多数生理过程,被公认为最重要的药物靶标。传统的GPCR靶向药物主要是结合正构位点的配体分子,随着研究不断深入,越来越多的GPCR被发现存在别构调控机制。别构调控可以增强受体的亚型选择性,降低药物毒副作用,因此靶向GPCRs的别构调控小分子设计和筛选是目前新药研发的前沿热点。然而存在于受体静态结构表面的潜在别构位点较少,而更多是在蛋白动态构象变化中短暂存在的隐式别构位点,其隐蔽性和瞬时性阻碍了别构调控药物的开发。
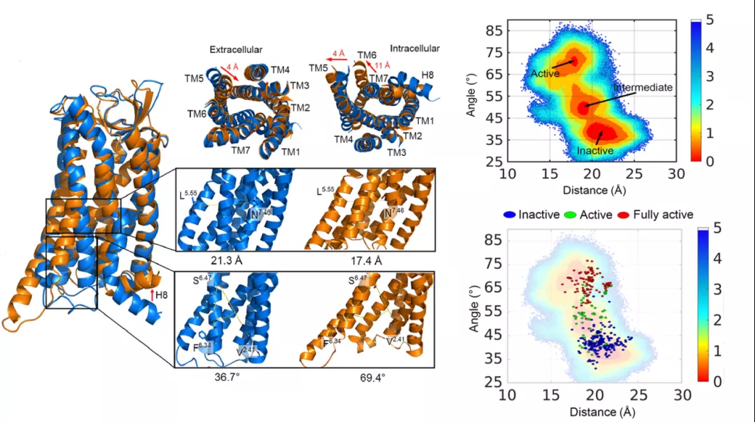
本研究团队以血管紧张素受体AT1R作为模式GPCR。AT1R是心血管和肾脏疾病的重要药物靶点,其过度激活会导致高血压和动脉瘤等疾病。拮抗AT1R的Gq和Arrestin活力分别对这两种疾病有显著疗效,据此已发展出临床的沙坦类药物。本研究从最近发表的AT1R失活态和激活态的晶体结构出发,识别了失活态和激活态之间的最小能量路径,绘制了AT1R激活过程的势能面,获得了稳定中间态,利用马尔科夫模型揭示了构象态之间转化的动力学性质,揭示了构象转变过程中关键氨基酸的微转换变化,为隐式别构位点的发现提供了基础。预测并验证了位于TM1-TM7和H8之间的P6区域是中间态独有的潜在别构位点。该位点在激活过程中随着H8的上移而出现,能够协同调控G蛋白和β-arrestin信号,说明其在AT1R受体构象变化中的重要作用。本工作发展了识别隐式别构机制及位点的新策略,为AT1R别构激动剂设计提供了新的思路。
孙金鹏教授研究团队在GPCR配体识别和信号转导机制领域连续取得进展,先后提出GPCR磷酸化编码的笛子模型理论(Nat Commun. 2015),揭示了磷酸化编码别构调控SH3蛋白的多聚脯氨酸码头分选机制和时序作用机制(Nat Chem Biol. 2018;Nat. Commun. 2021);和金沙9001w以诚为本入口于晓教授合作发现了AT1R与TRPC3偶联的新机制以及与北京大学孔炜教授团队发现了同型半胱氨酸和软骨寡聚基质蛋白为AT1R的内源性配体,从而介导心血管事件(Nat Commun. 2017;Nature Commun. 2018;Cell Res. 2021);揭示了胆汁酸受体激活的独特机制以及GPR97感知糖皮质激素的机制(Nature. 2020; Nature. 2021)。本研究是这些系列工作的延伸进展,得到了国家杰出青年科学基金等多项基金的资助与支持。
文章链接:http://www.nature.com/articles/s41467-021-25020-9








